Abstract
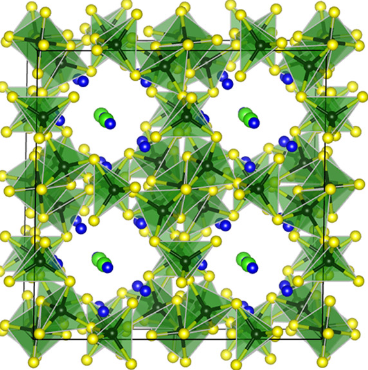
Motivated by previous investigations that showed very promising high ionic conductivities within rela- tively stable framework structures, we report a systematic first-principles study of the extended family of lithium (thio)boracites, consisting of eight chemical compositions. Three of the compositions—Li4B7O12Cl, Li4Al3B4O12Cl, and Li6B7S13Cl—are comparable to synthesized and analyzed materials reported in the exper- imental literature. The five additional compositions—Li4B7S12Cl, Li4Al3B4S12Cl, Li6B7O13Cl, Li6Al3B4O13Cl, and Li6Al3B4S13Cl—are predicted from the computational modeling and analysis presented in this paper. For each material, idealized ordered rhombohedral, cubic, or monoclinic ground-state structures are determined. Through various methodologies including thermodynamic, voltage window, and harmonic phonon analyses, stability is assessed for all eight Li (thio)boracite-derived compositions. Based on the dominant energetics of density functional theory, an analysis of the thermodynamically accessible phases predicts stability for Li4B7O12Cl only. The analysis of the voltage windows of these materials suggests that the sulfur materials are much more reactive in contact with lithium metal than their oxygen counterparts. This reactivity problem has been identified in other highly conducting solid electrolytes and various mitigation methods discussed in the literature look promising. Within the harmonic approximation, phonon analysis predicts that all eight materials are dynamically stable in their ground-state structures. Future investigations will focus on the performance of the family of lithium (thio)boracites, including ionic conductivity predictions and ion migration mechanisms.