Abstract
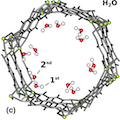
We calculate the carbon nuclear magnetic resonance (NMR) shielding for CO2 and the hydrogen shieldings for both H2 and H2O inside the metal organic framework MOF-74-Mg. Our ab initio calculations are at the density functional theory level using the van der Waals including density functional vdW-DF. The shieldings are obtained while placing the small molecules throughout the structure, including the calculated adsorption site for various loading scenarios. We then explore relationships between loading, rotational and positional characteristics, and the NMR shieldings for each adsorbate. Our NMR calculations show a change in the shielding depending on adsorbate, position, and loading in a range that is experimentally observable. We further provide a simple model for the energy and the NMR shieldings throughout the cavity of the MOF. By providing this mapping of shielding to position and loading for these adsorbates, we argue that NMR probes could be used to provide additional information about the position at which these small molecules bind within the MOF, as well as the loading of the adsorbed molecule.