Abstract
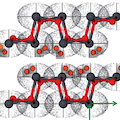
The structure, electronic, and dynamic properties of the two-layered α (litharge) and β (massicot) phases of PbO have been studied by density functional methods. The role of London dispersion interactions as leading component of the total interaction energy between layers has been addressed by using the Grimme’s approach in which new parameters for Pb and O atoms have been developed. Both gradient-corrected and hybrid functionals have been adopted using Gaussian-type basis sets of polarized triple-ζ quality for O atoms and small-core pseudopotential for the Pb atoms. Basis set superposition error (BSSE) has been accounted for by the Boys–Bernardi correction to compute the interlayer separation. Cross-checks with calculations adopting plane waves that are BSSE free have also been performed for both structures and vibrational frequencies. With the new set of proposed Grimme’s type parameters, structures and dynamical parameters for both PbO phases are in good agreement with experimental data.