Abstract
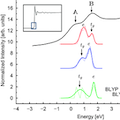
Comparison and prediction of the experimental XANES spectrum is a good measurement of the quality of the electronic structure calculations employed, and their ability to predict electronic transitions in solids. Here we present a comparison between BLYP+U and hybrid-BLYP calculations regarding the geometric, magnetic and electronic structures of α-Fe2O3 (hematite). Several values of U and different percentages of Fock-exchange have been screened to see how their contributions affect different properties of hematite, paying particular attention to the electronic structure. To estimate the quality of the various methods the calculated density-of- states were compared to the experimentally collected XANES spectrum of the iron K-edge, providing information about the orbitals describing the conduction band. We find that in agreement with previous studies DFT+U and hybrid-functional simulations can correctly predict the character of the valence band, but only Fock-exchange higher than 30% or U-values equal or larger than 6 eV properly reproduce the order between the tg and e orbitals in the conduction band, and can, therefore, be used to study and predict XANES spectra and electronic transitions in hematite.