Abstract
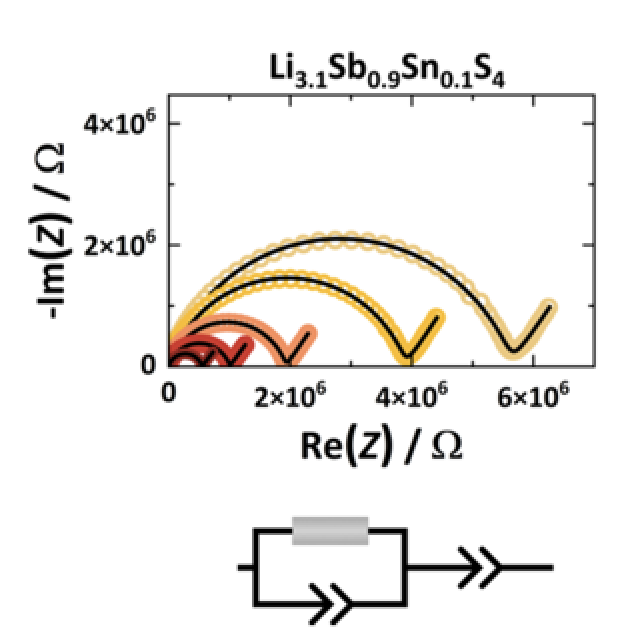
The search for highly conducting Li+ solid electrolytes focuses on sulfide- and halide-based materials, where typically the strongly atomic disordered materials are the most promising. The atomic disorder corresponds to flattened energy landscape having similar relative site energies for different Li+ positions facilitating motion. In addition, the highly disordered Li+ conductors have a negligible defect formation energy as moving charges are readily available. This work investigates the isovalent Li3Sb1-xPxS4 (0 ≤ x ≤ 0.5) and the aliovalent Li3+Sb1-xPxS4 (0 ≤ x ≤ 0.2) substitution series of thio-LISICON materials by using X-ray diffraction, high-resolution neutron diffraction, impedance spectroscopy and defect calculations. The starting composition Li3SbS4 has a low ionic conductivity of ~10−11 S∙cm−1 and both substituents improve the ionic conductivity strongly by up to four orders of magnitude. On the one hand, in substituted Li3SbS4 structures, only minor structural changes are observed which cannot sufficiently explain the significant impact on the Li+ conductivity. On the other hand, the Li+ carrier density reveals a correlation to the activation energy and first-principles defect calculations display a significantly reduced defect formation energy upon substitution. Here, we show within two different substitution series that the defect formation energy plays a major role for ionic motion in this class of thio-LISICON materials.